Raman spectra#
Introduction#
Raman spectroscopy probes vibrational modes that modulate the electric susceptibility \(\boldsymbol{\chi}\) of a material. The Raman intensity is proportional to the power spectral density of the time derivative of \(\boldsymbol{\chi}(t)\), evaluated along a molecular dynamics trajectory.
First-order Raman scattering involves a single phonon and is governed by crystal symmetry selection rules. BaZrO\(_3\) in its cubic phase (space group Pm\(\bar{3}\)m) is inactive for first-order Raman scattering; only second-order (two-phonon) scattering is allowed. Under pressure, BaZrO\(_3\) undergoes a cubic-to-tetragonal phase transition, which the NEP model used here, predicts to occur at approximately 14 to 16 GPa. In the tetragonal phase, first-order Raman becomes allowed and produces peaks that are considerably more intense than the second-order features.
TNEP models provide the susceptibility \(\boldsymbol{\chi}(t)\) at each MD step; get_raman_spectrum converts the autocorrelation function of \(\boldsymbol{\chi}(t)\) to the Raman spectrum. See the TNEP susceptibility tutorial for how to train the susceptibility model used here, and the TNEP polarization tutorial for the rank-1 (polarization) variant.
This tutorial is based on the work of Rosander et al., Phys. Rev. B 111, 064107 (2025) and Fransson et al., Chem. Mater. 36, 514 (2024). The TNEP formalism is described in Xu et al., J. Chem. Theory Comput. 20, 3273 (2024).
Simulation setup#
The simulations use a 20×20×20 supercell of the 5-atom cubic BaZrO\(_3\) primitive cell (40,000 atoms) and two NEP models:
nep_energy.txt— the PES (potential energy surface) model (NEP4).nep_susceptibility.txt— the rank-2 TNEP susceptibility model, identical to the model trained in the TNEP susceptibility tutorial.
The simulation protocol is:
NPT equilibration: 50,000 steps × 2 fs = 100 ps at 300 K and target pressure, using the stochastic cell-rescaling (SCR) barostat.
NVE production: 500,000 steps × 2 fs = 1 ns;
dump_polarizability 5writes \(\boldsymbol{\chi}(t)\) every 5 × 2 fs = 10 fs, giving 100,000 frames per run.
potential nep_energy.txt
potential nep_susceptibility.txt
time_step 2.0
velocity 300
ensemble npt_scr 300 300 100 0 0 0 80 80 80 100
dump_thermo 500
dump_restart 50000
run 50000
ensemble nve
dump_thermo 500
dump_polarizability 5
dump_restart 50000
run 500000
Simulations are run at pressures from 0 to 20 GPa to span the cubic-to-tetragonal phase transition. Each run takes approximately 30 minutes on a single NVIDIA GH200 GPU. Using the above settings you can run the MD yourself, but you can also download the pre-computed trajectories from Zenodo by running the next cell.
[1]:
import os
import zipfile
import urllib.request
url = 'https://zenodo.org/records/20679922/files/barium-zirconate-raman.zip'
if not os.path.exists('barium-zirconate-raman'):
urllib.request.urlretrieve(url, 'barium-zirconate-raman.zip')
with zipfile.ZipFile('barium-zirconate-raman.zip') as zf:
zf.extractall('.')
os.remove('barium-zirconate-raman.zip')
print('Downloaded and extracted barium-zirconate-raman/')
else:
print('barium-zirconate-raman/ already present')
barium-zirconate-raman/ already present
[2]:
from ase.io import read
basedir = 'barium-zirconate-raman'
atoms = read(f'{basedir}/300K-0GPa/model.xyz')
natoms = len(atoms)
print(f'{natoms} atoms ({natoms // 5} formula units BaZrO3)')
print(f'Cell: {atoms.cell.lengths().round(3)} Å')
40000 atoms (8000 formula units BaZrO3)
Cell: [84.18 84.174 84.196] Å
Raman spectrum at 300 K, 0 GPa#
We start with the cubic phase at ambient pressure. As BaZrO\(_3\) is Raman inactive in first order at 0 GPa, the spectrum is dominated by second-order (two-phonon) scattering.
The accessible frequency range is bounded at both ends. The lower cutoff is set by the NVE run length: the ACF is evaluated up to a maximum lag \(T_\mathrm{ACF}\) (by default 25% of the trajectory, giving 250 ps here), which limits the raw frequency resolution to \(\Delta\tilde{\nu} = 1/(c\,T_\mathrm{ACF}) \approx 0.13\,\mathrm{cm}^{-1}\). Gaussian window with t_sigma = 1 ps broadens spectral lines further to
\(\sigma_{\tilde{\nu}} = 1/(2\pi c\,t_\sigma) \approx 5\,\mathrm{cm}^{-1}\) (FWHM \(\approx\) 12 cm\(^{-1}\)), so features below approximately 25 cm\(^{-1}\) are not reliably resolved. Longer NVE runs lower this cutoff proportionally. The upper cutoff follows from the Nyquist criterion: \(\tilde{\nu}_\mathrm{max} = 1/(2\,c\,\Delta t)\) where \(\Delta t = 10\) fs gives \(\tilde{\nu}_\mathrm{max} \approx 1667\,\mathrm{cm}^{-1}\), well above the BaZrO\(_3\)
Raman features.
[3]:
from calorine.gpumd import read_polarizability
from calorine.tools import get_raman_spectrum
folder = f'{basedir}/300K-0GPa'
chi_df = read_polarizability(f'{folder}/polarizability.out', scale=natoms)
# dt = dump_polarizability × time_step = 5 × 2 fs = 0.01 ps
dt = 0.01 # ps
raman_df = get_raman_spectrum(
dt, chi_df, t_sigma=1.0,
polarization_in=[1, 0, 0], polarization_out=[1, 0, 0])
raman_df.head()
[3]:
| angular_frequency | wavenumber_invcm | raman_isotropic | raman_anisotropic | raman_polarized | |
|---|---|---|---|---|---|
| 0 | 0.000000 | 0.000000 | 2.696604e-20 | 8.806972e-20 | 4.080035e-20 |
| 1 | 0.012567 | 0.066714 | 2.696489e-20 | 8.806741e-20 | 4.079878e-20 |
| 2 | 0.025133 | 0.133428 | 2.696143e-20 | 8.806049e-20 | 4.079404e-20 |
| 3 | 0.037700 | 0.200142 | 2.695567e-20 | 8.804895e-20 | 4.078615e-20 |
| 4 | 0.050266 | 0.266857 | 2.694761e-20 | 8.803279e-20 | 4.077511e-20 |
Quantum corrections for second-order Raman#
Classical MD samples the Boltzmann distribution and underestimates spectral intensities at frequencies where \(\hbar\omega \gtrsim k_\mathrm{B}T\). For second-order scattering, the correction depends on the type of two-phonon process:
Overtone — two identical phonons each at \(\omega/2\):
\[f_\mathrm{ot}(\omega) = \left[\frac{\beta\hbar\omega/2}{1-e^{-\beta\hbar\omega/2}}\right]^2 \!\!(2 - e^{-\beta\hbar\omega/2})\]Combination band — two different phonons summing to \(\omega\):
\[f_\mathrm{cb}(\omega) = \left[\frac{\beta\hbar\omega/2}{1-e^{-\beta\hbar\omega/2}}\right]^2\]
These are applied via apply_quantum_correction with order='overtone' and order='combination'. First-order Raman (tetragonal phase) uses order='first' (standard single-phonon Bose-Einstein factor).
[4]:
from pandas import read_json
from calorine.tools import apply_quantum_correction
temperature = 300 # K
raman_cb = apply_quantum_correction(
raman_df, temperature, 'raman_polarized', order='combination')
wn = raman_df['wavenumber_invcm']
mask_700 = (wn > 600) & (wn < 800)
norm_factor = raman_cb['raman_polarized_qm'][mask_700].max()
def norm(s):
return s / norm_factor
# Experimental data (Toulouse et al., Phys. Rev. B 100, 134102 (2019))
expt = read_json(f'{basedir}/experimental-data-Toulouse2019-fig2b.json')
[5]:
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=(6, 4))
ax.plot(wn, norm(raman_df['raman_polarized']),
label='Classical')
ax.plot(wn, norm(raman_cb['raman_polarized_qm']),
label='Combination correction')
ax.plot(expt['frequency'], expt['intensity'],
'k--', label='Expt. Z(XX)Z (Toulouse 2019)')
ax.set_xlabel(r'Wavenumber (cm$^{-1}$)')
ax.set_ylabel('Z(XX)Z Raman intensity (normalized)')
ax.set_xlim(50, 1400)
ax.set_ylim(-0.05, 1.05)
ax.legend(frameon=False, fontsize='small')
fig.tight_layout()
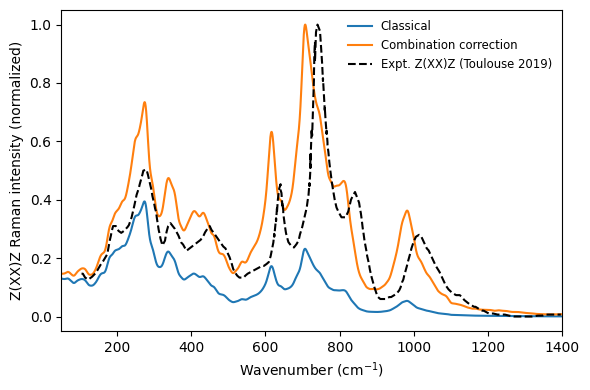
The overall agreement between the TNEP prediction and experiment is good. The broad second-order Raman envelope is reproduced in both shape and frequency range. The higher-frequency features above approximately 600 cm\(^{-1}\) are slightly redshifted relative to experiment, which could be a feature of the exchange-correlation functional used for the underlying density functional theory calculations or the classical treatment. The relative intensities of the low- and high-frequency regions differ slightly from experiment, which could also be caused to a small misalignment of the scattering geometry in the experiment. A detailed discussion is given in Rosander et al. (2025).
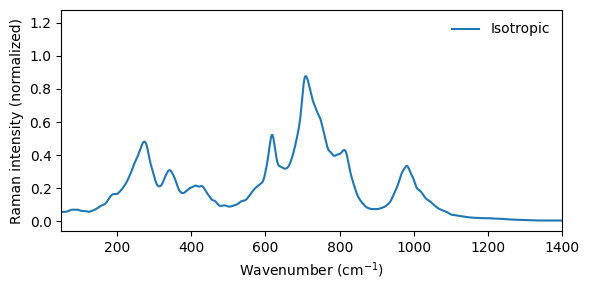
It is also possible to inspect the isotropic Raman spectrum.
[6]:
raman_iso_cb = apply_quantum_correction(
raman_df, temperature, 'raman_isotropic', order='combination')
fig, ax = plt.subplots(figsize=(6, 3))
ax.plot(wn,
raman_iso_cb['raman_isotropic_qm'] / norm_factor,
label='Isotropic')
ax.set_xlabel(r'Wavenumber (cm$^{-1}$)')
ax.set_ylabel('Raman intensity (normalized)')
ax.set_xlim(50, 1400)
ax.legend(frameon=False)
fig.tight_layout()
Pressure dependence#
We now compute the Raman spectrum at each pressure, track the structural phase transition via the lattice parameters, and compare the second-order (cubic) and first-order (tetragonal) spectra.
[7]:
import re
from glob import glob
from pandas import DataFrame
from calorine.gpumd import read_thermodynamic_data
dirs = glob(f'{basedir}/300K-*GPa')
results = []
for dname in dirs:
p_label = float(re.search(r'([\d.]+)GPa', dname).group(1))
thermo = read_thermodynamic_data(dname, normalize=True)
thermo = thermo[thermo.time > 0.2 * thermo.time.max()]
abc = sorted([
(thermo.alat / 20).mean(),
(thermo.blat / 20).mean(),
(thermo.clat / 20).mean(),
])
results.append({
'p_label': p_label,
'P': thermo.pressure.mean(),
'V': thermo.volume.mean(),
'a': abc[0],
'b': abc[1],
'c': abc[2],
})
averages = DataFrame(results).sort_values('P', ignore_index=True)
averages.head()
[7]:
| p_label | P | V | a | b | c | |
|---|---|---|---|---|---|---|
| 0 | 0.0 | 0.007108 | 14.921185 | 4.208935 | 4.209951 | 4.210406 |
| 1 | 4.0 | 3.977147 | 14.560038 | 4.175038 | 4.175129 | 4.176400 |
| 2 | 8.0 | 7.979239 | 14.237289 | 4.143982 | 4.144430 | 4.144906 |
| 3 | 12.0 | 11.993134 | 13.946974 | 4.115882 | 4.115891 | 4.116454 |
| 4 | 13.0 | 13.042885 | 13.875163 | 4.108634 | 4.108800 | 4.109563 |
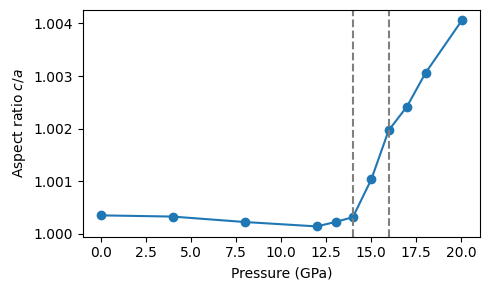
We first inspect the axial ratio as a function of pressure, which reveals the transition pressure. Here, we are satisfied with a rather approximate determination of the transition pressure, which occurs between 14 and 16 GPa. In production this analysis should be more carefully.
[8]:
fig, ax = plt.subplots(figsize=(5, 3))
ax.plot(averages['P'], averages['c'] / averages['a'], 'o-',)
ax.set_xlabel('Pressure (GPa)')
ax.set_ylabel('Aspect ratio $c/a$')
ax.axvline(14, ls='--', color='0.5')
ax.axvline(16, ls='--', color='0.5')
fig.tight_layout()
Now we compute the Raman spectra.
[9]:
spectra = {}
for dname in dirs:
p_label = float(
re.search(r'([\d.]+)GPa', dname).group(1))
chi = read_polarizability(f'{dname}/polarizability.out', scale=natoms)
df = get_raman_spectrum(
dt, chi, t_sigma=1.0,
polarization_in=[1, 0, 0], polarization_out=[1, 0, 0])
df = apply_quantum_correction(
df, temperature, 'raman_polarized', order='combination')
spectra[p_label] = df
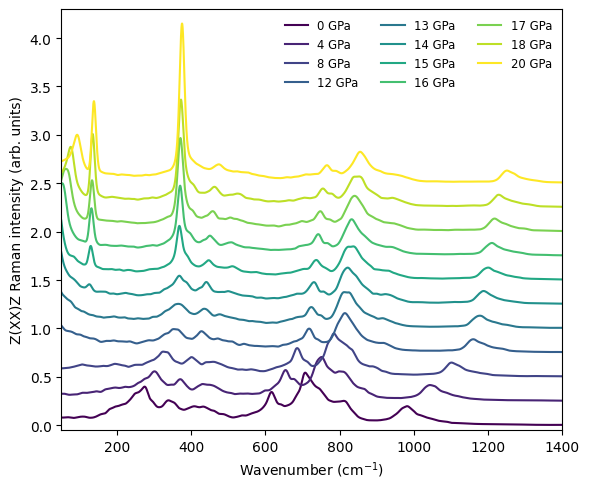
In the result below, we can see that as the pressure passes through the cubic-to-tetragonal phase transition around 14 to 16 GPa, the character of the Raman spectrum changes qualitatively. The broad, relatively weak second-order (two-phonon) scattering of the cubic phase is replaced by intense, narrow first-order peaks in the tetragonal phase. Because all spectra are normalized to a common reference (the maximum of the 0 GPa spectrum), this transition appears as a pronounced jump in absolute intensity in the waterfall plot.
[10]:
p0 = min(spectra)
norm_pres = spectra[p0]['raman_polarized_qm'].max()
offset = 0.25
n_p = len(dirs)
cmap = plt.colormaps['viridis'].resampled(n_p)
wn_all = next(iter(spectra.values()))['wavenumber_invcm']
fig, ax = plt.subplots(figsize=(6, 5))
for idx, (p_label, df) in enumerate(sorted(spectra.items())):
y = df['raman_polarized_qm'] / norm_pres + idx * offset
ax.plot(wn_all, y, color=cmap(idx), label=f'{p_label:.0f} GPa')
ax.set_xlabel(r'Wavenumber (cm$^{-1}$)')
ax.set_ylabel('Z(XX)Z Raman intensity (arb. units)')
ax.set_xlim(50, 1400)
ax.set_ylim(-0.05, 4.3)
ax.legend(frameon=False, fontsize='small', ncol=3)
fig.tight_layout()
Polarized Raman#
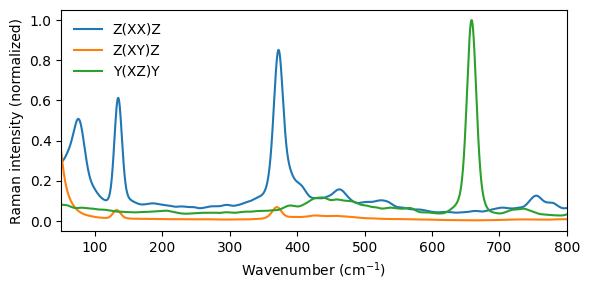
In an ordered crystal, Raman spectra can be recorded for specific scattering geometries, revealing the symmetry of individual phonon modes. The geometry is commonly expressed in Porto notation \(A(BC)D\): \(A\) is the propagation direction of the incoming light, \(B\) is the incoming polarization, \(C\) is the outgoing polarization, and \(D\) is the propagation direction of the scattered light.
get_raman_spectrum accepts optional polarization_in and polarization_out unit-vector arguments (corresponding to \(B\) and \(C\)); when both are specified, the function returns a raman_polarized column.
Following Rosander et al. (2025), Fig. 5, we compute three geometries for the tetragonal phase:
Z(XX)Z — both beams along \(z\), both polarized along \(x\) (parallel)
Z(XY)Z — both beams along \(z\), crossed polarizations (perpendicular)
Y(XZ)Y — beams along \(y\), incoming \(x\), outgoing \(z\)
The highest available pressure directory is used as a representative tetragonal-phase simulation (the cubic-to-tetragonal transition occurs at approximately 14 to 16 GPa).
[11]:
folder_tet = f'{basedir}/300K-18GPa'
pol_tet = read_polarizability(f'{folder_tet}/polarizability.out', scale=natoms)
geometries = {
'Z(XX)Z': ([1, 0, 0], [1, 0, 0]),
'Z(XY)Z': ([1, 0, 0], [0, 1, 0]),
'Y(XZ)Y': ([1, 0, 0], [0, 0, 1]),
}
pol_spectra = {}
for name, (e_in, e_out) in geometries.items():
df = get_raman_spectrum(
dt, pol_tet, t_sigma=1.0,
polarization_in=e_in,
polarization_out=e_out,
)
df = apply_quantum_correction(df, temperature, 'raman_polarized', order='first')
pol_spectra[name] = df
[12]:
fig, ax = plt.subplots(figsize=(6, 3))
for name, df in pol_spectra.items():
wn_p = df['wavenumber_invcm']
y = df['raman_polarized_qm']
ax.plot(wn_p, y / y.max(), label=name)
ax.set_xlabel(r'Wavenumber (cm$^{-1}$)')
ax.set_ylabel('Raman intensity (normalized)')
ax.set_xlim(50, 800)
ax.legend(frameon=False)
fig.tight_layout()
Discussion#
Second-order vs first-order Raman: at 0 GPa the cubic phase produces a broad second-order (two-phonon) spectrum; above the cubic-to-tetragonal transition, first-order Raman peaks emerge and are considerably more intense. The lattice-parameter plot shows the onset of the phase transition through the divergence of the \(a\) and \(c\) parameters.
Quantum corrections: without quantum corrections, classical MD underestimates spectral intensities at high frequencies where \(\hbar\omega \gg k_\mathrm{B}T\). The overtone and combination corrections differ primarily at higher frequencies: the overtone correction falls off more slowly, and the two limiting cases bracket the experimental spectrum, reflecting that the real second-order spectrum is a mixture of both types of processes.
Polarized Raman: different scattering geometries probe different irreducible representations of the crystal point group. In the tetragonal phase, only modes of specific symmetry contribute to each geometry, making polarized Raman a powerful tool for mode assignment.
A detailed discussion of the computed spectra and their comparison to experiment can be found in Rosander et al., Phys. Rev. B 111, 064107 (2025), and Fransson et al., Chem. Mater. 36, 514 (2024). Experimental Raman spectra for BaZrO\(_3\) at ambient conditions are from Toulouse et al., Phys. Rev. B 100, 134102 (2019).