Dielectric functions#
This tutorial demonstrates how to compute the dielectric function tensor \(\epsilon^{\alpha\beta}(\omega) = \epsilon_1^{\alpha\beta}(\omega) + i \epsilon_2^{\alpha\beta}(\omega)\) from a GPUMD molecular-dynamics simulation. GPUMD writes the time derivative of the polarization \(\dot{\mathbf{P}}(t)\) to a file named dpdt.out when using a qNEP model. The imaginary part of the dielectric tensor \(\epsilon_2^{\alpha\beta}(\omega)\) is
related to the power spectral density of \(\dot{P}_\alpha(t)\) via
where \(V\) is the simulation cell volume, \(T\) is the temperature, and \(\epsilon_0\) is the permittivity of free space. The isotropic average \(\bar{\epsilon}_2(\omega) = \frac{1}{3}\sum_\alpha \epsilon_2^{\alpha\alpha}(\omega)\) reduces to the scalar dielectric function for cubic or isotropic systems. The real part \(\epsilon_1^{\alpha\beta}(\omega)\) is obtained component-wise via the Kramers-Kronig relation
The example data are from qNEP simulations of BaTiO\(_3\) at 100, 331, and 450 K from Z. Fan et al., J. Chem. Theory Comput. 22, 4787 (2026).
[1]:
base = 'https://zenodo.org/records/20648097/files'
for T in [100, 331, 450]:
fname = f'dpdt-BaTiO3-{T}K.txt'
import os, urllib.request
if not os.path.exists(fname):
urllib.request.urlretrieve(f'{base}/{fname}', fname)
print(f'Downloaded {fname}')
else:
print(f'{fname} already present')
dpdt-BaTiO3-100K.txt already present
dpdt-BaTiO3-331K.txt already present
dpdt-BaTiO3-450K.txt already present
Reading the data#
The read_dpdt function reads a dpdt.out file into a pandas.DataFrame. The columns dPx, dPy, dPz are the Cartesian components of \(\dot{\mathbf{P}}(t)\) in units of \(e\)Å/fs, and Px, Py, Pz are the corresponding polarization components in units of \(e\)Å. The time step is read directly from the time column as the difference between consecutive rows.
[2]:
from calorine.gpumd import read_dpdt
temperatures = [100, 331, 450] # K
dfs_raw = {T: read_dpdt(f'dpdt-BaTiO3-{T}K.txt') for T in temperatures}
dfs_raw[450].head()
[2]:
| time | dPx | dPy | dPz | Px | Py | Pz | |
|---|---|---|---|---|---|---|---|
| 0 | 0.02 | -3.06855 | -1.117540 | 1.867190 | -61.3709 | -22.3507 | 37.3438 |
| 1 | 0.04 | -5.94163 | 0.009431 | 1.536860 | -180.2030 | -22.1621 | 68.0809 |
| 2 | 0.06 | -5.86222 | -2.869390 | 0.291382 | -297.4480 | -79.5499 | 73.9085 |
| 3 | 0.08 | -4.57303 | -4.939870 | 0.963120 | -388.9080 | -178.3470 | 93.1709 |
| 4 | 0.10 | -1.08819 | -5.468230 | -0.305113 | -410.6720 | -287.7120 | 87.0687 |
Computing the dielectric function#
get_dielectric_function computes both parts of the dielectric tensor from all three Cartesian components of \(\dot{\mathbf{P}}(t)\), returning the six unique Voigt components of the imaginary part \(\epsilon_2^{\alpha\beta}(\omega)\) together with the real part \(\epsilon_1^{\alpha\beta}(\omega)\) obtained via the Kramers-Kronig relation. The required physical parameters are
volume: the simulation cell volume in Å\(^3\),temperature: the temperature in K.
The keyword column='dP' (the default) selects the dPx/dPy/dPz columns from read_dpdt. The time step is extracted automatically from the time column. The optional parameter t_sigma sets the width of a Gaussian window applied to the autocorrelation function before the Fourier transform (in ps); smaller values produce smoother spectra while larger values yield sharper features.
[3]:
%%time
import numpy as np
from calorine.tools import get_dielectric_function
t_sigma = 2 # Gaussian window width in ps; smaller values give smoother spectra
lattice = np.array([[81.047018, 0.023688, -0.017573],
[ 0.014659, 80.976750, -0.016819],
[-0.017772, -0.026703, 80.939036]])
volume = np.abs(np.linalg.det(lattice)) # Angstrom^3
print(f'Cell volume: {volume:.1f} Angstrom^3')
dfs = {}
for T in temperatures:
dfs[T] = get_dielectric_function(dfs_raw[T], volume, T, column='dP', t_sigma=t_sigma)
dfs[450].head()
Cell volume: 531196.7 Angstrom^3
CPU times: user 1min 47s, sys: 14.8 s, total: 2min 2s
Wall time: 1min
[3]:
| angular_frequency | wavenumber_invcm | epsilon_imag_xx | epsilon_imag_yy | epsilon_imag_zz | epsilon_imag_yz | epsilon_imag_xz | epsilon_imag_xy | conductivity_xx | conductivity_yy | conductivity_zz | conductivity_yz | conductivity_xz | conductivity_xy | epsilon_real_xx | epsilon_real_yy | epsilon_real_zz | epsilon_real_yz | epsilon_real_xz | epsilon_real_xy | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0.006283 | 0.033357 | 124255.284323 | 59899.173855 | 41507.546044 | -1965.406627 | -6210.587144 | 7150.681630 | 6912.770709 | 3332.407606 | 2309.214856 | -109.342677 | -345.517417 | 397.818272 | 62862.620438 | 31549.732382 | 22417.644882 | -946.858042 | -2967.715355 | 3434.593445 |
| 1 | 0.012567 | 0.066714 | 62149.270547 | 29962.758741 | 20763.426580 | -982.586187 | -3105.680122 | 3575.548379 | 6915.177239 | 3333.873197 | 2310.288981 | -109.329644 | -345.560427 | 397.841367 | 31555.872535 | 16458.496943 | 11960.202017 | -451.554422 | -1402.762191 | 1632.702157 |
| 2 | 0.018850 | 0.100071 | 41456.876064 | 19989.807161 | 13853.010279 | -654.927121 | -2070.882673 | 2383.929419 | 6919.187705 | 3336.315735 | 2312.079141 | -109.307891 | -345.632071 | 397.879838 | 15076.692173 | 8514.590987 | 6455.453043 | -190.875112 | -579.063694 | 684.318563 |
| 3 | 0.025133 | 0.133428 | 31117.883584 | 15007.720701 | 10401.019451 | -491.058181 | -1553.612344 | 1788.188890 | 6924.801479 | 3339.735051 | 2314.585267 | -109.277368 | -345.732287 | 397.933652 | 9885.493452 | 6012.157721 | 4721.399918 | -108.756631 | -319.587462 | 385.573541 |
| 4 | 0.031417 | 0.166785 | 24920.248748 | 12021.979431 | 8332.398456 | -392.705044 | -1243.352559 | 1430.799575 | 6932.017682 | 3344.130904 | 2317.807258 | -109.238007 | -345.860991 | 398.002767 | 7561.505183 | 4891.890736 | 3945.128978 | -71.994153 | -203.428301 | 251.844760 |
Kramers-Kronig integration method#
The Kramers-Kronig step is performed internally by get_dielectric_function (controlled by return_real_part, which defaults to True). Two integration methods are available via the kk_method keyword:
'vectorized'(default): exact trapezoid rule, \(\mathcal{O}(n^2)\) time and memory.'fft': \(\mathcal{O}(n \log n)\) Hilbert-transform approximation; faster for large arrays but can deviate near sharp features.
The section below compares both methods on the 100 K data, which has the sharpest phonon peaks and provides the most stringent test.
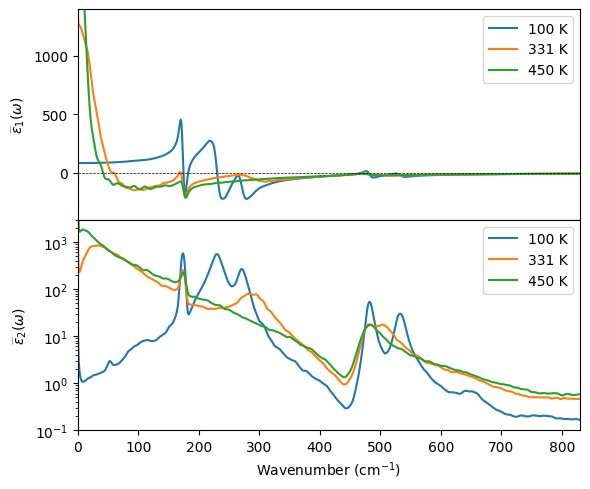
Visualization#
[4]:
import matplotlib.pyplot as plt
def isotropic_average(df, prefix):
return (df[f'{prefix}_xx'] + df[f'{prefix}_yy'] + df[f'{prefix}_zz']) / 3
fig, (ax1, ax2) = plt.subplots(2, 1, figsize=(6, 5), sharex=True)
for T in temperatures:
df = dfs[T]
ax1.plot(df.wavenumber_invcm, isotropic_average(df, 'epsilon_real'), label=f'{T} K')
ax2.plot(df.wavenumber_invcm, isotropic_average(df, 'epsilon_imag'), label=f'{T} K')
ax1.axhline(0, color='k', linewidth=0.5, linestyle='--')
ax1.set_ylabel(r'$\widebar{\epsilon}_1(\omega)$')
ax1.legend()
ax1.set_xlim(0, 830)
ax1.set_ylim(-400, 1400)
ax2.set_xlabel(r'Wavenumber (cm$^{-1}$)')
ax2.set_ylabel(r'$\widebar{\epsilon}_2(\omega)$')
ax2.legend()
ax2.set_yscale('log')
ax2.set_ylim(0.1, 3e3)
fig.tight_layout()
fig.subplots_adjust(hspace=0)
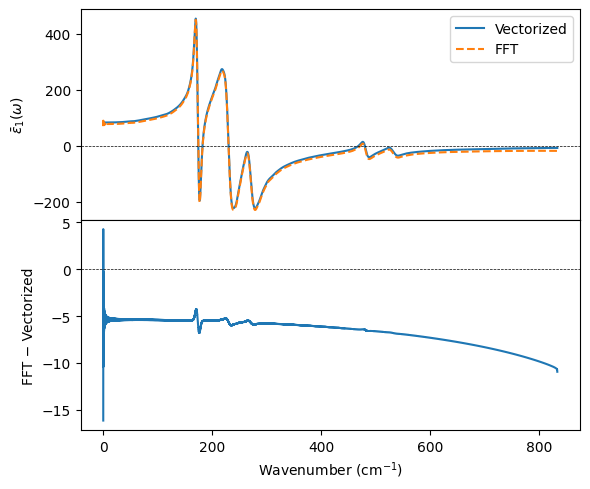
Accuracy: FFT vs. vectorized#
dfs[100] was computed with the default kk_method='vectorized'. The cell below recomputes the 100 K spectrum with kk_method='fft' for comparison. The two methods agree well across most of the spectrum, but the FFT approximation can deviate near sharp features and at larger frequencies.
[5]:
df_vec = dfs[100].iloc[5:]
df_fft = get_dielectric_function(dfs_raw[100], volume, 100,
column='dP', t_sigma=t_sigma, kk_method='fft').iloc[5:]
fig, (ax1, ax2) = plt.subplots(2, 1, figsize=(6, 5), sharex=True)
ax1.plot(df_vec.wavenumber_invcm, isotropic_average(df_vec, 'epsilon_real'), label='Vectorized')
ax1.plot(df_fft.wavenumber_invcm, isotropic_average(df_fft, 'epsilon_real'), '--', label='FFT')
ax1.axhline(0, color='k', linewidth=0.5, linestyle='--')
ax1.set_ylabel(r'$\bar{\epsilon}_1(\omega)$')
ax1.legend()
diff = isotropic_average(df_fft, 'epsilon_real').to_numpy() - isotropic_average(df_vec, 'epsilon_real').to_numpy()
ax2.plot(df_vec.wavenumber_invcm, diff)
ax2.axhline(0, color='k', linewidth=0.5, linestyle='--')
ax2.set_xlabel(r'Wavenumber (cm$^{-1}$)')
ax2.set_ylabel('FFT − Vectorized')
fig.tight_layout()
fig.subplots_adjust(hspace=0)
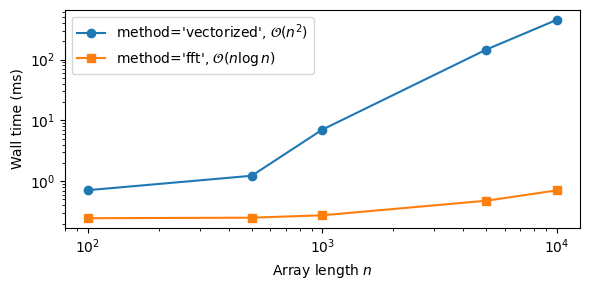
Kramers-Kronig performance#
The timing comparison below illustrates the performance advantage of the 'fft' method over the 'vectorized' approach, which comes at a small cost in accuracy (see above).
[6]:
from calorine.tools import apply_kramers_kronig
import time
from pandas import DataFrame
sizes = [100, 500, 1000, 5000, 10000]
times_vec = []
times_fft = []
for n in sizes:
df_test = DataFrame({'angular_frequency': np.linspace(0.1, 100.0, n),
'epsilon_imag': np.sin(np.linspace(0.1, 100.0, n))})
nrep = max(3, 200 // n)
t0 = time.perf_counter()
for _ in range(nrep):
apply_kramers_kronig(df_test, method='vectorized')
times_vec.append((time.perf_counter() - t0) / nrep * 1000)
t0 = time.perf_counter()
for _ in range(500):
apply_kramers_kronig(df_test, method='fft')
times_fft.append((time.perf_counter() - t0) / 500 * 1000)
fig, ax = plt.subplots(figsize=(6, 3))
ax.loglog(sizes, times_vec, 'o-', label=r"method='vectorized', $\mathcal{O}(n^2)$")
ax.loglog(sizes, times_fft, 's-', label=r"method='fft', $\mathcal{O}(n \log n)$")
ax.set_xlabel('Array length $n$')
ax.set_ylabel('Wall time (ms)')
ax.legend()
fig.tight_layout()